Aula Virtual 03
HACER PARA APRENDER
HACER PARA APRENDER
Como el objetivo será conocer la cantidad total de un determinado amplicón (ya se refiera a un gen, una bacteria, etc.), es imprescindible contar con unas muestras de cantidad conocida de dicho amplicón para poder usarlas de referencia.
Para el diseño y la preparación de la curva patrón se requiere una muestra con una cantidad exacta conocida de moléculas. Esta muestra puede ser:
– Un fragmento de ADN de doble cadena o de cadena sencilla
– Un cDNA
– Un producto amplificado por PCR de la secuencia del ADN de interés clonado en un plásmido
– Un producto de la PCR convencional
– La síntesis directa de la secuencia del ADN blanco
En general, el estándar absoluto externo es medido como número de copias por microlitro de muestra. La cuantificación de la secuencia de interés suele ser correspondiente a la cantidad del organismo de interés (patógenos), a la cantidad de copias de un gen extraño (OMG), etc.
Para diseñar la curva patrón, en número de copias por microlitro de muestra, se recurrirá a la técnica más sencilla para la obtención de un ADN de alta calidad y se determinará la cantidad de ADN de la muestra gracias al peso medio de un par de bases de ADN (que está establecido en 330 Da. para cadena simple y 660 Da. para doble cadena). Será necesario también conocer el peso del ADN de la muestra (típicamente establecido por determinación de absorbancia a 260 nm) y conocer el número de Avogadro que nos permite convertir los moles presentes en la muestra en número de moléculas (1 mol = 6,023 x 1023 moléculas).
| En un ejemplo |
|
Si se amplifica un fragmento de 800 pares de bases por PCR y se purifica, el peso de cada mol de fragmentos de ADN en la muestra será de: 800 p.b. x 660 g/mol = 528.000 g/mol Como 1 mol son 6,023 x 1023 moléculas, por el número de Avogadro: 528.000 g/mol x 1 mol/6,023 x 1023 = 8,48 x 10-19 g Si se parte de un stock de 20 ng/µl: si una molécula pesa 8,48 x 10-19 g (8,48 x 10-10 ng), en 20 ng/µl habrá: 20 ng/µl / 8,48 x 10-10 ng = 2,37 x 1010 moléculas de ADN/µl Es decir, en cada microlitro de la muestra estándar hay más de 20.000 millones de moléculas. Con ese dato ya se pueden hacer diluciones seriadas para construir la recta patrón: Co x Vo = Cf x Vf Cf = 109 moléculas/ µl Se llevaría a cabo una primera dilución a 109 moléculas y, de ahí, se pueden establecer las diluciones necesarias para construir la curva patrón. Por ejemplo, de 108, 107, 106, 105, 104, 103 y 102.
|
Así, conociendo la longitud del fragmento de ADN presente en la muestra (plásmido + inserto; secuencia sencilla; secuencia de doble cadena…), podrá calcularse el peso de cada una de las moléculas de ADN presentes en dicha muestra.
Con este dato y conociendo la cantidad de ADN presente en la muestra en ng/µl podremos calcular el número de moléculas de ADN presentes en la muestra convirtiéndolo mediante el uso del número de Avogadro.
Conociendo la cantidad de moléculas en la muestra inicial se llevarán a cabo las diluciones necesarias para cubrir el rango de posibles concentraciones en las muestras “problema”. Una vez realizadas las diluciones se llevará a cabo un primer experimento introduciendo entre 5 y 7 puntos para determinar el rango apropiado para que los diferentes puntos de la curva patrón tengan su Ct entre los ciclos 16 y 35.
También la cantidad inicial de cada muestra tendrá que ser la suficiente para que la detección de la señal fluorescente se realice entre los ciclos 20 y 35, realizando posteriormente los cálculos matemáticos necesarios para establecer la cantidad total del ADN amplificado en la muestra inicial.
Lo más frecuente en los experimentos de tiempo real es utilizar placas de 96 pocillos, aunque hay disponibles equipos para placas de 48 pocillos y para placas de 384 pocillos. La elección de la placa dependerá del equipo disponible.
En todos los experimentos de cuantificación absoluta por tiempo real son necesarias una serie de muestras control que es necesario conocer para el diseño del experimento:
– Recta patrón: es necesario incorporar muestras control de cantidades conocidas, con, al menos, tres puntos, para la realización de una recta aceptable.
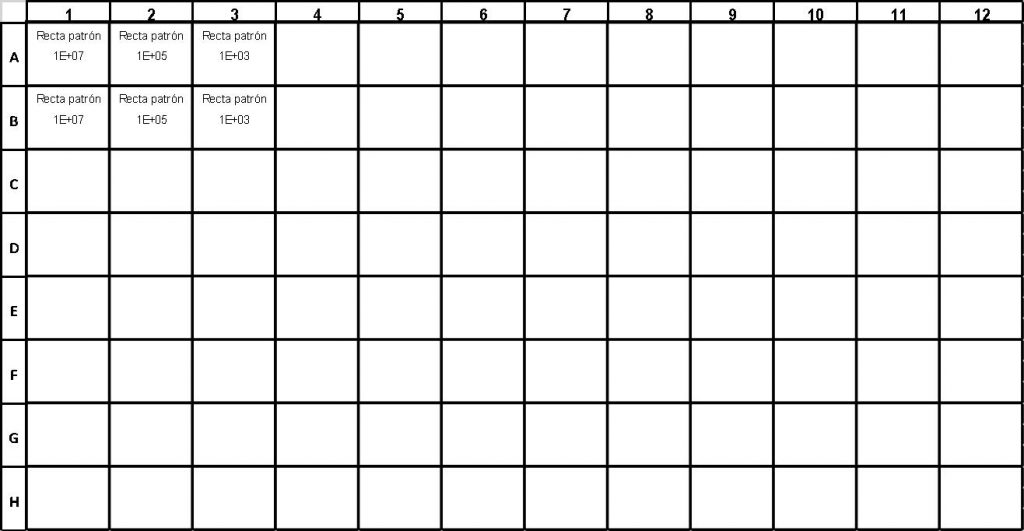
– Control negativo de reacción: se incorpora una muestra en la que estén todos los reactivos excepto el ADN para comprobar que no hay contaminación en ningún reactivo que nos esté dando datos falsos. Es habitual denominarlo NTC (del inglés, No Template Control).
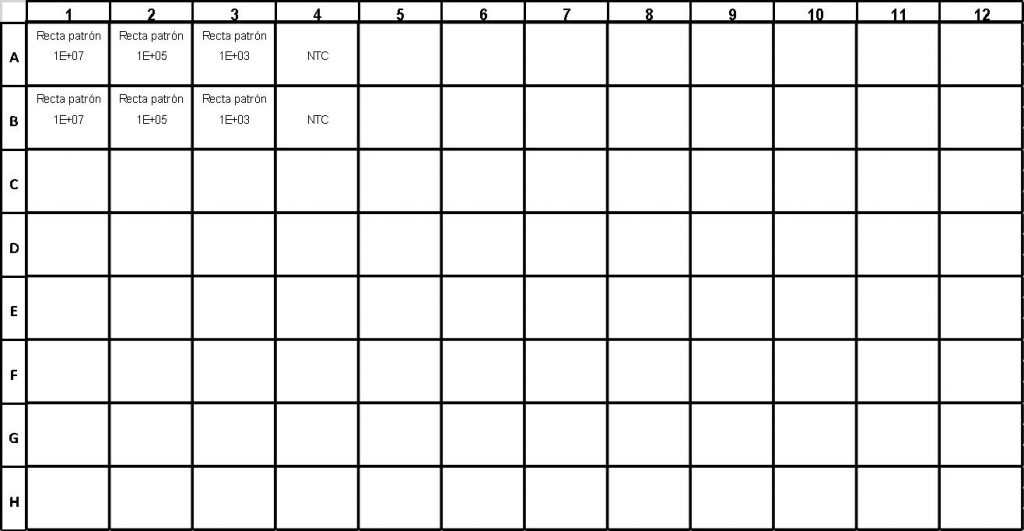
– Control positivo de reacción: las propias muestras de la curva patrón son controles positivos, ya que son muestras en las que sabemos que SI hay ADN y deben amplificar.
– Muestras a analizar: es importante realizar un diseño experimental previo a los análisis cuantitativos para determinar qué información quiero obtener y qué muestras pueden proporcionármelo.
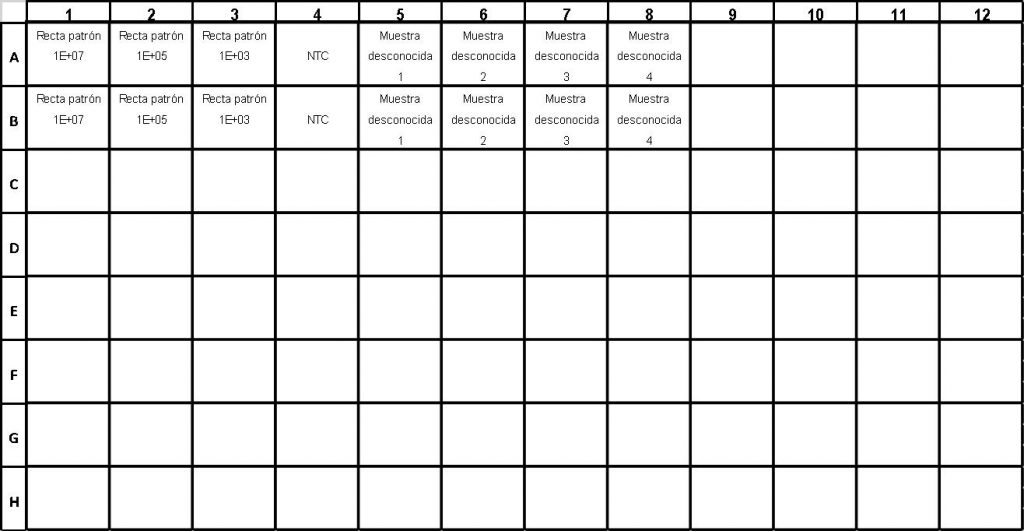
– Réplicas técnicas: es recomendable que, de todas las muestras, se realicen triplicados o, al menos, duplicados, para poder minimizar distorsiones en los resultados como resultado de errores de pipeteo.
– Réplicas biológicas: para estudios de I+D siempre es necesario la obtención de muestras equivalentes en las mismas condiciones. Es recomendable realizar experimentos en diferentes momentos y placas para comparar entre ellos y, en cada placa, referidos a su propia recta patrón. NUNCA se analizarán resultados en función de una recta patrón de otra placa, ya que las diferencias en la emisión o recepción de fluorescencia entre experimentos pueden alterar sustancialmente los resultados.
– Genes u organismos a analizar: en cada placa podrán analizarse varias sondas o parejas de primers para diferentes genes, organismos, etc. (por ejemplo para analizar la presencia de varios patógenos en las muestras). Para ello SIEMPRE será necesaria una curva patrón por cada pareja de primers o sonda específica, que parta de una muestra estándar de cantidad conocida para esa secuencia. En la placa ejemplo, cada color representa el análisis de un gen diferente.
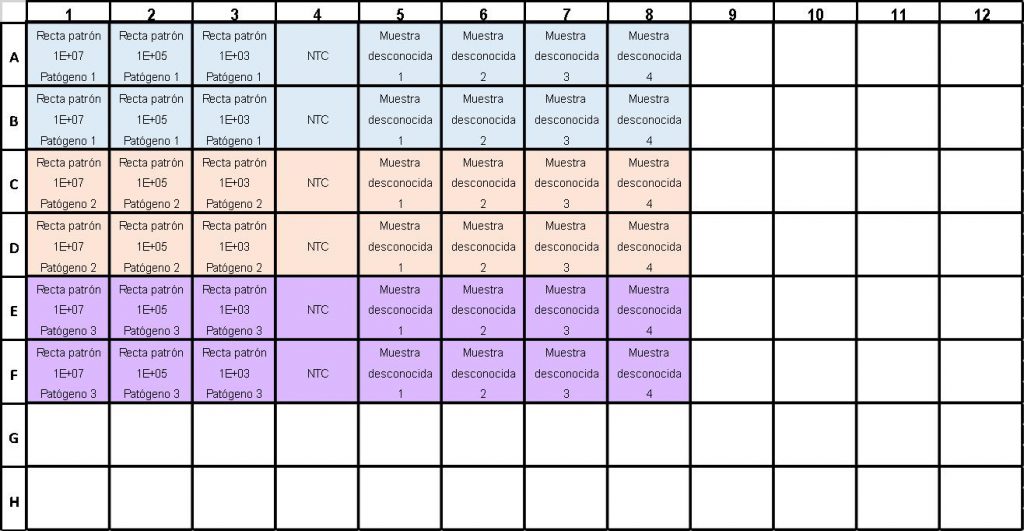
En la placa del ejemplo se está analizando la presencia de tres posibles patógenos en cuatro muestras diferentes.
Las condiciones de la PCR van a depender de las temperaturas de melting de los primers/sondas, de la polimerasa que se utilice y del equipo de PCR a tiempo real.
Las condiciones generales, que son las que suele tener el equipo por defecto y para las que están optimizadas la mayoría de mix y sondas comerciales consisten en un primer ciclo de activación de la polimerasa de 10 minutos a 95°C, y entre 35 y 40 ciclos de amplificación (con 10 segundos de desnaturalización a 95°C y 30 segundos de anillamiento a 60°C). La PCR a tiempo real no suele incluir etapas de extensión a 72°C, los amplicones que se generan son de pequeño tamaño por lo que es suficiente la rampa entre 60 y 95°C para que se lleve a cabo la extensión.
Cuando la química seleccionada es un agente intercalante es conveniente realizar una curva de melting al final de la amplificación para confirmar que las amplificaciones han sido específicas.
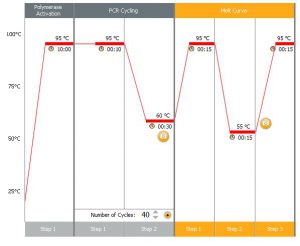
Los equipos de PCR a tiempo real recogen los datos de fluorescencia a lo largo de la amplificación y, una vez concluida, ofrecen los resultados en base a la curva patrón y la información aportada en la plantilla.
Las gráficas de amplificación muestran la fluorescencia detectada a lo largo de los ciclos que se han realizado (generalmente 35-40). En los primeros ciclos de amplificación no hay cambios significativos en la señal de fluorescencia y se define un rango que se considera fluorescencia basal; se establece así lo que se denomina la línea basal. El software genera una gráfica de amplificación en la que se ha sustraído la línea basal. Para ello, calcula una tendencia matemática de la señal fluorescente normalizada (valores Rn, de fluorescencia, correspondientes a los ciclos de la línea basal) y, a continuación, un algoritmo busca el punto de la gráfica de amplificación en el que la señal fluorescente normalizada con la línea basal (valor delta Rn [ΔRn]) cruza el umbral. El ciclo de amplificación en el que el valor ΔRn cruza el umbral se define como Ct (o Cq) y es en el que se basan los cálculos para determinar los resultados del experimento.
En un experimento de cuantificación absoluta el equipo indicará, directamente, el número total de copias (ng/μL, μmol/ μL o equivalentes genómicos…) presentes en la muestra analizada, calculadas a partir de los datos que le hemos dado para la elaboración de la curva patrón.
Es muy importante tener en cuenta que el número de copias que indica el equipo dependen de la recta patrón y los datos que se le hayan proporcionado. Lo más habitual es indicar el número de copias en la propia muestra, es decir, el total de copias del amplicón que se han puesto inicialmente en cada uno de los pocillos. De esta manera, si se han utilizado 2 µl de la dilución: millón / microlitro, en la recta habremos indicado dos millones de copias (uno por cada microlitro que hemos utilizado). Por tanto, los resultados también estarán indicados como total en cada pocillo y será necesario hacer los cálculos que correspondan para determinar el total en la muestra de partida.
| En un ejemplo |
|
Si se están analizando unas muestras de alimentos para determinar la presencia de determinadas bacterias. Se extrae el ADN presente en 1 gramo de alimento y queremos saber cuántas bacterias de cada tipo hay en ese gramo. Datos que tenemos que conocer: – El volumen final en el que se ha extraído el ADN total de la muestra: en el ejemplo 100 µl. – Volumen de dicho ADN extraído que vamos a añadir en cada pocillo: en el ejemplo 2 µl. En ese caso será necesario dividir el resultado que ofrece el equipo entre 2 (para obtener el número de copias por microlitro). Si el resultado que ofrece el equipo es: 283.544,47 283.544,46 copias / 2 = 141.772,23 copias por microlitro Por tanto en cada uno de los microlitros del ADN total extraído hay 141.772,23 copias copias de esa bacteria. Para determinar el total en la muestra completa será necesario multiplicar por 100, ya que es el volumen en el que está extraído el ADN: 141.772,23 x 100 = 14.177.223 bacterias de la especie X Como sabemos que en esos 100 µl está el ADN presente en un gramo de la comida que se está estudiando, el resultado de nuestro estudio concluiría que: Hay 14.177.223 bacterias de la especie X por cada gramo de la comida estudiada |
Para dar por válidos estos resultados es necesario que los umbrales de calidad y los errores técnicos estén limitados.
– Error de pipeteo: para confirmar que en los pocillos se ha añadido la misma cantidad de ADN total inicial y que las variaciones entre pocillos son debidas a diferencias reales y no a errores de pipeteo debemos comparar entre los pocillos de cada duplicado o triplicado. La desviación estándar entre los datos de Ct de los dos o tres pocillos debe ser menor de 0,3.
– Recta patrón: una recta patrón confiable es aquella en la que los diferentes puntos de la recta se desvían poco de la recta estándar generada a partir de los mismos. Esta desviación se indica mediante el R2. Y, para que se trate de una recta confiable y, por tanto, la extrapolación de los puntos sea veraz el R2 debe ser mayor a 0,95.
– Extrapolación: es aconsejable que los puntos de nuestras muestras queden incluidos dentro de los puntos de la recta patrón analizada. Esto proporciona mayor exactitud en los resultados.
* Material adicional: hay disponibles varios artículos de ejemplos del uso de la PCR a tiempo real para cuantifiación absoluta en diferentes áreas.